A platform for engineering CAR-T cells with enhanced cancer-killing ability and reduced toxicity. By redesigning the transmembrane domain of the CAR using a computational protein design platform, researchers created synthetic receptors that form stable, specific oligomeric structures. These modifications improve cytotoxic efficacy while significantly lowering the risk of inflammatory cytokine release associated with cytokine storms.
- CAR-T platforms for safer and more effective cancer therapy
- Synthetic receptor design for additional cell-based therapies
- Enhanced cytotoxicity against target cancer cells
- Reduced inflammatory cytokine release
- Modular integration into existing CAR designs
- De novo-designed transmembrane domains allow precise receptor tuning
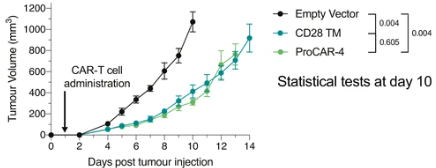
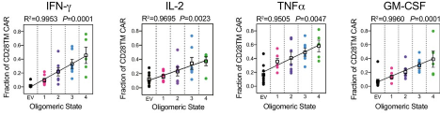
proCAR-4 matches CD28TM CAR tumor control in vivo with substantially lower cytokine release in vitro
Top: MC38 tumor growth
Bottom: In vitro cytokine production of programmed CARS (proCAR) is substantially lower compared to the commonly used CD28 TMD, and scales with the proCAR oligomeric state.
The computational design platform is established. Proof-of-concept has been validated in mouse CAR-T models targeting HER2+ tumors, showing increased cytotoxicity and reduced cytokine release. Further testing in human T cells and additional CAR backgrounds is planned.

