A new class of small molecules offers a targeted strategy for treating Huntington’s disease (HD) and related trinucleotide repeat expansion disorders (TREDs). These compounds bind one of the interaction surfaces between transcription factor Spt5 and RNA Polymerase II—specifically required for expressing expanded, pathogenic repeats—thereby selectively silencing the mutant allele while preserving normal gene expression. This precision mechanism may reduce toxicity and improve therapeutic outcomes.
- Treatment of Huntington’s Disease (HD)
- Potential treatment of other TREDs, including: Spinocerebellar Ataxias 1, 2, 3 and 7, , Myotonic Dystrophy Type 1 etc.
- Allele-selective inhibition: targets mutant but not normal gene expression
- Novel mechanism: bind the surfacebetween transcription factor Spt5 and RNA Polymerase II
- Demonstrated in vitro and in vivo efficacy
- No apparent toxicity
Hit compounds have been identified and validated in vitro, demonstrating selective activity. In vivo studies in HD mouse models indicate a favorable safety profile, reduced anxiety, and improved motor skills. Further compound optimization is ongoing, including testing in Spinocerebellar Ataxias models.
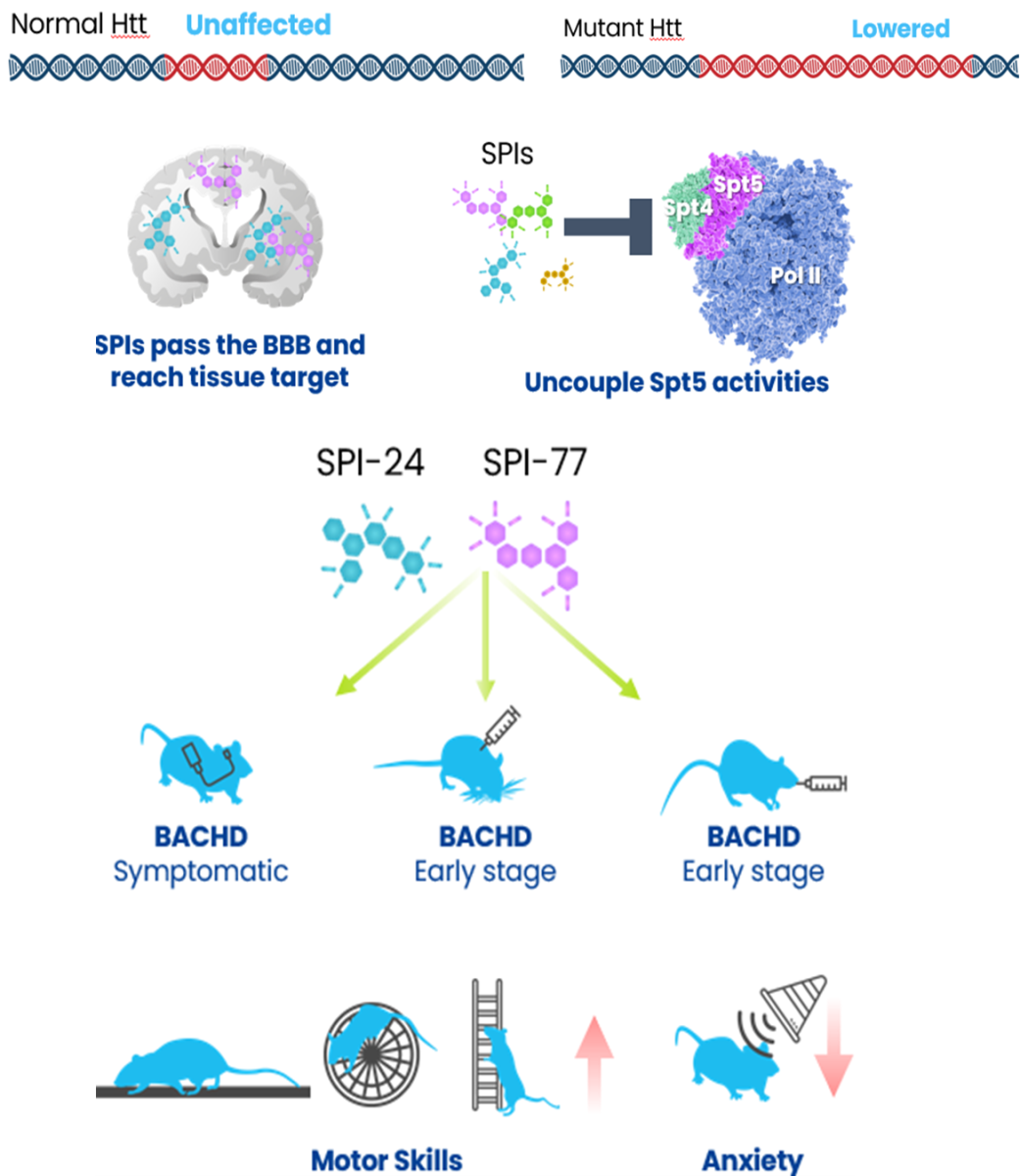
Administration of SPI-24 and SPI-77 directly, subcutaneously, and orally to symptomatic and early-stage BACHD HD mouse model. These SPIs pass the BBB, selectively lower mutant Htt expression, restore BDNF levels and mitochondrial function, improve motor and anxious-like symptoms, and delay disease progression.

