18646
Overview
Single-cell RNA sequencing (scRNA-seq) methods enable the detection of only 10%-15% of the cellular transcriptome, rendering low-expressing genes in the dark. WRAP-seq (Well-based RNA Amplification and Pooling) combined with TRAP-seq (Targeted RNA Amplification and Pooling) enable the detection of targeted sets of low-expressing genes of interest on top of the whole transcriptome. These gene sets can be tailored to provide in-depth analysis of specific biological pathways.
Applications and Differentiation
Applications
- Dissecting cell subpopulations
- Deciphering cell state within the tumor microenvironment
- TCR/BCR analysis linked with the whole transcriptome
- Identifying specific isoforms
- Tailored applications supported by ‘Traps’ – panels for targeted genes of interest
Differentiation
- Single-cell sequencing of targeted genes of interest in parallel to whole transcriptomics analysis
- Superior sensitivity compared to commonly used methods
- High-throughput and cost-effective
Development Stage
- WRAP-seq is fully developed
- TRAP-seq has a POC of several target genes, and is in validation stages for additional targets
- A proof-of-concept study demonstrated superior sensitivity compared to a standard scRNA-seq method.
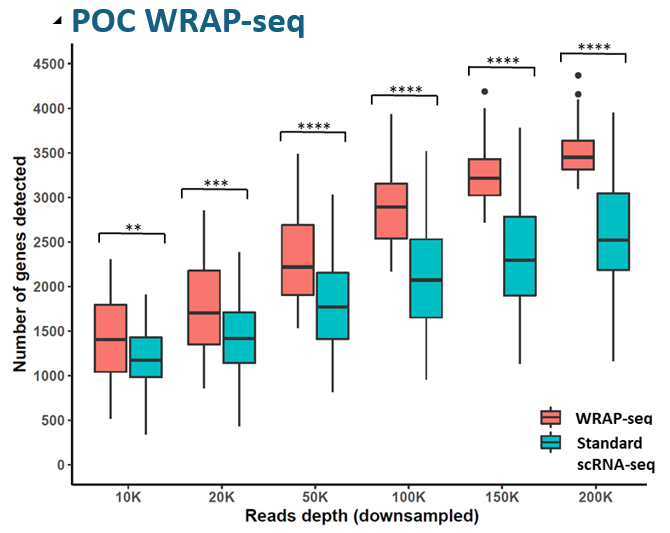
Compared with data from Mereu et al., Nature Biotechnology 2020
Patent Status:
PCT Published: Publication Number: WO 2023/199311 A1

Contact for more information
